
Total de visitas: 103732
|
POLIPOSE ADEMATOSA
Relato de Caso:
Polipose adenomatosa de cólon e Síndrome de Gardner: relato de caso clínico
Introdução
Síndrome é um conjunto de sinais e sintomas que caracterizam uma entidade patológica. A polipose múltipla do cólon (PMC) é uma doença genética localizada em cromossomos autossômicos e portanto, de herança autossômica dominante. Nos indivíduos afetados há desenvolvimento de pólipos colorretais e estes assumem grande risco de desenvolvimento de câncer com o passar dos anos. O gene responsável por esta doença e o APC (do inglês, adenomatous polyposis coli), que está localizado no braço longo do cromossoma 5 (5q)1. A Síndrome de Gardner é uma variante da PMC no qual os indivíduos também desenvolvem tumores extra-intestinais, em particular osteomas e tumores desmóides2. Existe uma forma atenuada da doença (AFAP) associada com menos pólipos, mas ainda com alto risco para câncer colorretal. Desta forma é nosso objetivo relatar a condução de um caso de polipose familiar, que com a elaboração do diagnóstico definitivo de uma descendente (filha), é que realmente concluiu-se se tratar de síndrome de Gardner, uma variação fenotípica da polipose adenomatosa familial, uma vez que o progenitor já havia falecido de neoplasia intestinal sem ter pesquisado a possibilidade desta variação. Cabe ressaltar que foi obtido termo de consentimento livre e esclarecido com a mãe da paciente, e o Comitê de Ética em Pesquisa da Universidade de Santa Cruz do Sul aprovou a forma desta apresentação científica.
REVISÃO DE LITERATURA
Ballhausen (2000) conceitua a polipose múltipla do cólon como sendo uma síndrome de herança dominante, com predisposição ao câncer colorretal. Leblanc (2000) considera a PMC como sendo uma patologia associada a tumores malignos do sistema nervoso central, predominantemente meduloblastomas e glioblastoma multiformes (síndrome de Turcot); e com osteomas craniofaciais ou síndrome de Gardner. A designação Síndrome de Gardner (SG) é usada para variantes fenotípicas de PMC, com sintomas extracólicos adicionais3. Desta forma a SG seria variante da síndrome de Turcot no qual meduloblastoma predominam, associadas com uma mutação do gene da polipose adenomatosa de cólon4. Tal diferença é percebida pela demonstração de tumores benignos não-neurogliais presentes nos pacientes com o SG. A caracterização molecular deveria revelar se os tumores intracranianos benignos podem ser representados por uma manifestação pleiotrópica do gene APC ou se seriam implicados outros genes5. Beuschlein et al. (2000), quando se referem a PMC são enfáticos na afirmação de que os pólipos intestinais sofrem transformação maligna na vida adulta. A SG, por sua vez seria uma variante de PMC, havendo tumores desmóides, osteomas e lesões pigmentadas da retina, concomitantemente com manifestações intestinais6. Trata-se de um distúrbio raro, de caráter hereditário relacionado a gene autossômico dominante, com penetração incompleta e expressividade variada7,8. Ocorre mutação no gene supressor de tumor6,8,9. Se não detectada ou sem tratamento, todos os pacientes desenvolvem carcinoma de cólon numa idade muito jovem. A descoberta cedo pode ser difícil, por causa de fenótipos atenuados ou mutações espontâneas.
RELATO DO CASO CLÍNICO
Paciente F.S.S., sexo feminino, 13 anos apresenta-se para consulta proctológica com queixa principal de freqüente sangramento durante as excreções e com diarréia persistente. Em sua história médica pregressa, havia retirado cirurgicamente uma lesão em pele da fronte e outra em couro cabeludo, que segundo relato da mãe tratava-se de lesão cística e ambas foram diagnosticadas como sendo o mesmo tipo de lesão, apenas com localização diferente. Sua história familiar reporta, que o pai morreu aos 27 anos de adenocarcinoma de intestino, bem como três tias, uma prima e avó (conforme heredograma na Figura 1). Foi proposto exame colonoscópico que apresentou múltiplos pólipos por toda extensão do intestino grosso (Figura 2), sendo realizada polipectomia que indicou, histopatologicamente, displasia leve, com trânsito intestinal normal. Houve a necessidade de endoscopia digestiva alta, que apresentou o estômago tomado por lesões polipóides (Figura 3). Submeteu-se logo em seguida à tomografia computadorizada do segmento cefálico que indicou a presença de lesões hiperdensas sugestivas de osteomas em etmóide, esfenóide, maxila e mandíbula (Figuras 4, 5 e 6, seqüencialmente). Em 2001 a paciente submeteu-se à remoção profilática do intestino grosso, com reconstrução através de bolsa ileal tipo Utsonomya, com excelente evolução pós-operatória. No exame histopatológico não houve a presença de lesão maligna dos pólipos analisados. Foram removidos ainda os dentes 15, 25 e 45, em retenção intra-óssea nos maxilares, juntamente com tecido ósseo circunjacente, que histopatologicamente comprovou tratar-se de osteoma. As lesões em etmóide e esfenóide não foram exploradas cirurgicamente, propondo-se acompanhamento radiográfico. A paciente utiliza inibidor de COX-2 (Celecoxib) permanentemente e submete-se a consultas periódicas para acompanhamento há 4 anos. A função intestinal encontra-se equilibrada, com duas evacuações por dia, sem quadros hematológicos compatíveis com anemia.
DISCUSSÃO
Com a descoberta dos genes responsáveis pela polipose familial múltipla de cólon (APC) e o subtipo manifesto como síndrome de Gardner, o diagnóstico pode ser precoce, com instituição de tratamento preventivo à neoplasia maligna colorretal. A mutação gênica do APC no códon 1309 antecipa em torno de 10 anos o aparecimento do câncer3. Investigação complementar de outros órgãos é importante, pois existem evidências que a perda da heterozigosidade do gene da polipose (APC) seria o responsável pela tumorigênese colorretal e também vinculado ao aparecimento de outras neoplasias em outros sítios anatômicos5,10. Na região do corpo gástrico podem ocorrer pólipos das glândulas, que raramente desenvolvem-se na ausência de pólipos intestinais, sendo uma das manifestações extracólon freqüentes da SG10. Tal informação é que leva à investigação endoscópica da via digestiva alta e quando alteração poliposa estiver presente, o tratamento está indicado com o uso de antiinflamatório (indometacina)11.
A glândula tiróide também deve ser observada em pacientes com PMC, pois apresenta relação com neoplasia nesta glândula endócrina2,12. Os tumores desmóides podem ser complicação séria em pacientes com SG, pois o seu aparecimento dentro da cavidade abdominal é raro13,14. Trata-se de uma neoplasia benigna que apresenta invasão local fibromatosa e, depois da excisão cirúrgica, tende a recorrer periodicamente1,15. Cerca de 8% dos pacientes com SG apresenta complicações advindas dos tumores desmóides, como obstrução uretral e fistulização, uma vez que o ritmo de crescimento é acelerado7.
Outra manifestação extraintestinal da SG são as lesões cutâneas4. Histologicamente se assemelham a fibromas, consistindo em pacotes de colágeno espessos, sem uma organização estrutural, entre os quais são encontrados fibroblastos ocasionalmente. As margens freqüentemente apresentam estruturas circunvizinhas que incluem gordura adjacente, músculo e nervo9.
No tecido ósseo os osteomas é que fazem parte da SG, podendo ser surpreendidos em diferentes ossos do viscero e neurocrânio2. São massas com densidade homogênea da periferia à base, com margens lisas ou suavemente lobuladas e sem um tecido mole acompanhando12. A partir do diagnóstico definitivo e dependendo da localização pode-se realizar seguimento clínico ou excisão parcial, pois não há risco de recidiva ou metástases12. Quando comprometem a função, como limitação de abertura bucal, opta-se pela remoção cirúrgica.

Figura 1. Heredograma demonstrando herança autossômica dominante.
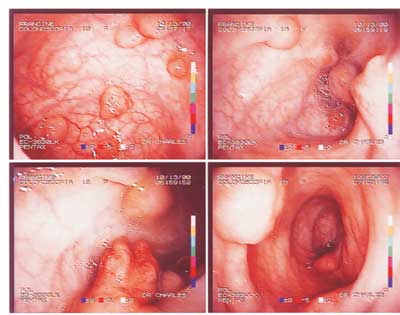
Figura 2. Colonoscopia diagnóstica, demonstrando várias imagens poliposas no intestino.

Figura 3. Endoscopia digestiva, demonstrando vários pólipos em corpo e fundo gástrico (esquerda); antro e piloro (direita).
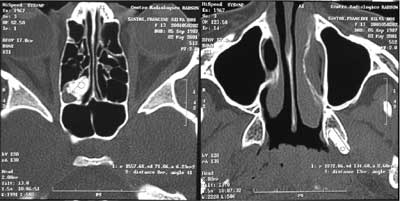
Figura 4. Tomografia computadorizada (TC) demonstrando em etmóide (esquerda) e entre as lâminas do processo pterigóide do esfenóide (direita), imagens compatíveis com osteomas.
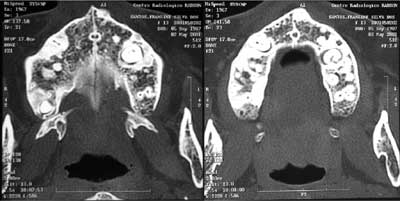
Figura 5. TC demonstrando dentes retidos (pré-molares) na maxila, circundados por uma imagem hiperdensa, compatível com osteomas em ambos os lados.

Figura 6. TC demonstrando dente retido na mandíbula (pré-molar direito) circundado por osso mais denso (esquerda); já no lado direito da mandíbula apenas condensação óssea. Ambas imagens compatíveis com osteomas.
CONSIDERAÇÕES FINAIS
O trabalho multidisciplinar na área da saúde é extremamente importante no que diz respeito a patologias que apresentam envolvimento de múltiplos órgãos. É com este intuito que a presença de dentes retidos associados com imagens radiográficas radiopacas sugestivas de osteomas nos ossos maxilares chamem a atenção dos profissionais da área da Odontologia para possibilidade da síndrome de Gardner5. Muitas vezes os pacientes encontram-se em investigação Proctológica, pela presença da polipose intestinal, necessitando de confirmação diagnóstica pelos achados radiográficos bucomaxilofaciais. A preocupação deve ser redobrada nos pacientes jovens, pois a SG é uma patologia perigosa. Apesar da possibilidade de diagnóstico endoscópico precoce, a intervenção cirúrgica radical (colectomia total profilática) é bem indicada, melhorando sobremaneira a qualidade e expectativa de vida dos pacientes com diagnóstico de síndrome de Gardner. Há também grande possibilidade destes pacientes desenvolverem outros vários tumores de origem mesenquimal, razão pela qual devem estar sempre em proservação, ou seja, o acompanhamento é fundamental. O aconselhamento genético é importante, pois esclarece ao paciente e seus familiares as reais possibilidades de descendentes apresentarem os problemas de seus pais.
|
|
