
Total de visitas: 103735
|
SÍNDROME DE EDWARDS
A Síndrome de Edwards

O primeiro caso de trissomia 18 foi descrito por John H. Edwards, no ano de 1960. A trissomia do 18 é a segunda mais frequentes síndrome de trissomia autossômica, com incidência estimada em 1:3500 a 1:7000 em nascidos vivos; e predominância no sexo feminino na razão de 3:1. Provavelmente 95% dos fetos com trissomia do 18 são abortados espontaneamente.
Cariótipo
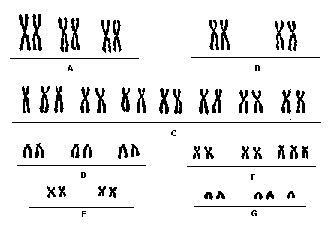
A sobrevivência pós-natal também é pequena, sendo estipulada desde poucos dias até alguns meses. Apesar de a etiopatogenia ser desconhecida, verifica-se uma nítida associação entre a ocorrência da síndrome e a idade materna avançada decorrida da não-disjunção meiótica do cromossomo 18, a maioria dos pacientes apresentam uma trissomia regular, com cariótipo 47XX + 18.
Dentre os restantes, cerca da metade é constituída por casos de mosaicismos, apresentando genótipo 46XX/ 47XX + 18; sendo que, esses desenvolvem manifestações menos graves, sobrevivem por mais tempo e nascem de mães jovens. Estudos recentemente realizados, demonstram que na maior parte dos casos ( 85% ), o erro ocorre na disjunção cromossômica da meiose materna, e somente 15% na meiose paterna.

As manifestações da trissomia 18 sempre incluem retardamento mental e atraso do crescimento e, às vezes malformações graves no coração. O crânio é excessivamente alongado na região occipital. O pavilhão das orelhas é dismórfico, com sulcos. A boca é pequena. O pescoço é curto. Há uma grande distância intermamilar. Os genitais externos são anômalos. O dedo indicador é maior do que os outros e flexionado sobre o dedo médio. Os pés têm as pontas arqueadas. As unhas costumam ser hipoplásticas.
Estudos anatomo- patológico e de imagem, realizados no Hospital Infantil Pequeno Príncipe entre janeiro de 89 a janeiro de 99, permitem a identificação de um grande número de malformações de órgãos, podendo comprometer praticamente todos os sistemas do organismo. As malformações do sistema cardiovascular ocorrem em 80% das crianças com trissomia 18. Contudo, nesse levantamento, somente 58% dos lactentes apresentavam algum tipo de anormalidade cardíaca, sendo todos submetidos a estudo ecocardiográfico. As malformações cardíacas encontradas foram múltiplas, mantendo relação fiel com a literatura internacional, sendo a comunicação interventricular a mais frequente ( 64% ), seguida de comunicação interatrial (57% ) e anomalias valvares com frequencia variável. A ocorrência da comunicação interventricular nesse estudo mostrou-se pouco abaixo da relatada na literatura ( em torno de 75-100% dos casos de trissomia 18 ) 3,8. A dextroposição da aorta é uma alteração raramente observada na trissomia 18, sendo que um dos pacientes estudados apresentou tetralogia de Fallot.

No sistema gastrointestinal, as malformações ocorrem em aproximadamente 80% dos casos, sendo o divertículo de Meckel e a atresia do esôfago as mais frequentemente relatadas 2,7,10. Dos 14 pacientes estudados nessa pesquisa, apenas 14% apresentavam divertículo de Meckel e 7% atresia de esôfago com fístula traqueoesofágica. O volvo do sigmóide, também frequentemente descrito, ocorreu em 21% das crianças estudadas e em 35% foi firmado diagnóstico de refluxo gastrioesofágico. Somente um dos pacientes apresentou estenose hipertrófica de piloro, em concordância com a baixa frequência dos relatos internacionais.
As malformações do sistema genito-urinário ocorrem em aproximadamente 60% dos portadores de trissomia 18. Nesse levantamento, tais malformações foram observadas em 58% dos casos. As mais frequentes foram criptorquidia ( 37% ), rim policístico ( 28% ), refluxo vesico-uretral ( 7% ) e cisto renal ( 7% ). Rins em ferradura e persistência da lobulação renal, usualmente descritos, não foram observados em nenhuma das crianças estudadas.
Cerca de 30% dos portadores da trissomia 18 demonstram algum comprometimento do SNC, sendo os mais frequentes: alterações do padrão dos giros cerebrais, alterações morfológicas cerebelais, mielomeningocele, anomalias do corpo caloso e hidrocefalia. Nesse levantamento, 42% dos pacientes apresentavam atrofia cerebral de graus variados, demonstrada na tomografia axial computadorizada de crânio, 7% agenesia do corpo caloso e 14% mielomeningocele.
Atualmente, há forte tendência que todas as crianças com aspectos clínicos compatíveis com a Síndrome de Edwards realizem estudo genético mais precoce possível. Os autores do presente trabalho acreditam que a confirmação diagnóstica da trissomia 18 é de suma importância, para posterior aconselhamento genético e avaliação criteriosa da realização de procedimentos invasivos de alto risco em um recém-nascido de prognóstico reservado.

Características:
Deficiência mental e crescimento
Hipertonicidade
Implantação baixa das orelhas
Mandíbula Recuada
Rim duplo
Ocorrência 1/6.000 nascimentos
5% a 10% sobrevive o 1º ano
|
|

